我要投稿
我要投稿2017年2月3日,美国能源部劳伦斯伯克利国家实验室宣布,其研究人员首次建立并训练机器自主学习算法,来高精度地预测某些金属间化合物的缺陷。相关论文名为《通过融合初始建模和机器学习,预测B2金属间缺陷行为》发表在了《npj计算材料学》期刊上。
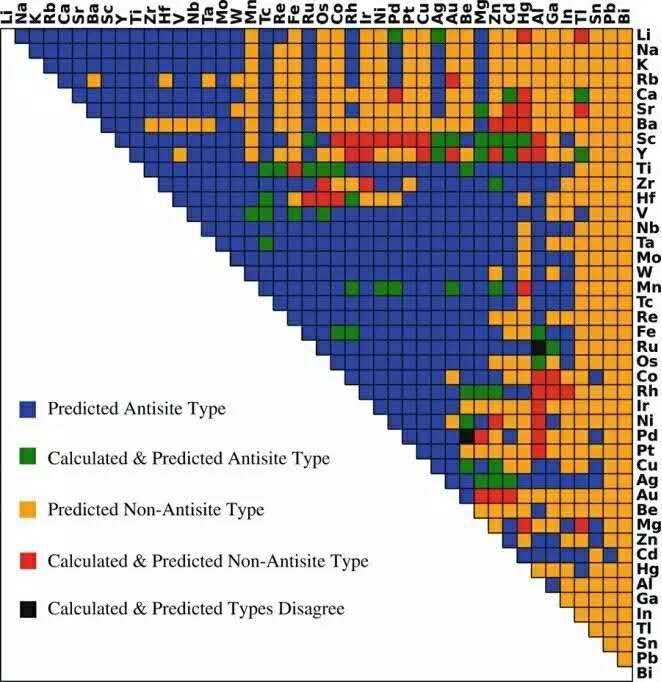
图:对B2金属间化合物主要缺陷类型进行预测的结果。颜色显示了预测与计算结果的关系(论文截图,劳伦斯伯克利国家实验室图片)
这种方法将会促进对于应用在汽车、航空航天以及其他更多领域中的新型先进合金和轻质新材料的研究。材料在化学上永远不是绝对纯净的,在结构上也永远不是完美无瑕的。它们几乎永远存在缺陷,这些缺陷会对材料的性质产生很大的影响。这些缺陷可能表现为某种“空缺”,即在材料的基本晶体结构中存在“孔洞”等,或错位缺陷,如基本原子位于错误的晶体位点上。了解上述的这些材料缺陷对于设计材料的科学家来说是至关重要的,因为这些缺陷对材料内部的结构和强度有着长期的影响。
一般来说,研究人员主要采用被称为密度函数计算的定量计算方法,来预测某种材料中会形成怎样缺陷,以及这些缺陷如何影响材料的性质。尽管是有效的,但这种方法对于应用于点缺陷的计算成本非常昂贵,这限制了这种方法的使用范围。“密度函数计算在一个小型单元模型上应用是非常有效的,但当你想让你的建模单元更大,所需要的计算能力也要大大增加。”前任伯克利实验室博士后、本篇论文的第一作者巴拉特·梅达塞尼(Bharat Medasani)解释道。“并且由于在单一的材料中计算建模缺陷的成本太高,对于成千上万种材料进行这强行的建模是不可行的”。
为了克服这些计算上的挑战,梅达塞尼和他的同事们研发并训练了一种可自主学习算法的机器,来预测金属间化合物的点缺陷,并致力广泛应用于观察B2晶体结构。起初,他们从材料项目数据库中选择了100个这些化合物的样本,然后在位于美国国家能源研究科学计算中心的超级计算机上进行密度函数计算(该中心隶属实验室内的科学用户设施办公室),从而识别缺陷。因为他们只有很小数据量大样本来计算,梅达塞尼和他的团队采用称为梯度提升的统计法来提高机器学习方法的准确性。在这种方法中,机器学习的额外模型会被顺序建立,并且会与之前建立的模型相结合,从而使得模型预测与密度函数计算的差异最小化。
这项工作是首次在预测材料点缺陷时应用机器自主学习模型,其最高的预测准确性可达75%以上。
文章链接:
Bharat Medasani, et al, "Predicting defect behavior in B2 intermetallics by merging ab initio modeling and machine learning," npj Computational Materials 2, Article number: 1 (2016), doi:10.1038/s41524-016-0001-z
(本文来源:中国航空工业发展研究中心,作者:陈济桁;)
如若转载,请注明e科网。
如果你有好文章想发表or科研成果想展示推广,可以联系我们或免费注册拥有自己的主页
- 机器学习
- 材料缺陷










 赞 2
赞 2
 回复
回复